Introduction
Hearing loss is one of the most common sensorineural disorder and affects one out of 500 live births. The prevalence of hearing loss increases as children get old.1) While many causes such as viral infections, ototoxic drugs and acoustic trauma could induce hearing loss, more than half of congenital bilateral profound deafness cases has been estimated to be attributed to genetic cause.2,3)
As considerable portion of genetic hearing loss is monogenic, the deafness phenotype could be predicted relatively accurately from genetic diagnosis. Identification of genetic cause can provide valuable information without further expensive testing.4) First, the prognosis of hearing, whether hearing loss will be worsen or not, could be predicted. Second, knowing the cause of hearing loss will allow prediction of the efficacy of certain therapeutic approaches and optimal intervention could be performed. For example, in genetic hearing loss with mutations of specific genes, such as, GJB2, SLC26A4, COCH, OTOF, and MYH9, mitochondrial mutations, cochlear implantation is supposed to lead to successful results.5) Third, the possibility that future children or other family members would have hearing loss could be estimated.5,6,7) Fourth, identification of mutations of hearing loss genes could detect the specific cause of a children's hearing loss, which cannot be identified by Universal Newborn Hearing Screening.4) From this, in children, to whom the linguistic development is crucial, early intervention could be performed.8) Therefore, genetic testing has become an important component for diagnosis of congenital hearing loss from etiologic and prognostic perspectives. Recently, as next-generation sequencing has been used commonly, the genetic diagnosis could be performed with high-throughput sequence analysis. We developed new diagnostic pipeline with next-generation sequencing. This paper will describe the result of our new diagnostic pipeline combining phenotype-driven candidate gene approach and targeted exome sequencing (TES) to find out the causative mutation of hearing loss.
Various Application of Next-Generation Sequencing (NGS) in Deafness
Genetic etiology of sensorineural hearing loss (SNHL) is extraordinarily heterogeneous. Therefore, to identify causative mu-tation in each patient, all the candidate genes should be screened. Before the era of next-generation sequencing, screening all the candidate genes was impossible. The previous method detecting the disease causing mutation was sequencing specific candidate gene and it was limited to the genes related to a certain phenotypic marker, such as GJB2, SLC26A4, or OTOF. This method could find out the causative mutation in only 10-20% of familial nonsyndromic hearing loss (NSHL). It was impossible to idetify the causative mutation in NSHL cases which were not related to a certain phenotypic marker. Therefore there has been a need to develop a new method to find out causative mutation in the patient with hearing loss. This need could be satisfied with next-generation sequencing (NGS). Studies for finding the causative mutation in hearing loss patients have grown rapidly with the advancement of NGS technique. NGS allowed whole genome, whole exome, and targeted gene sequencing to be more feasible and it leaded to easier identification of the causative mutation.4) However, whole genome sequencing is still too expensive and too much compared to other sequencing. While, whole exome sequencing includes just exome, which is protein coding regions (exons) and constitute 1% of the human genome. Eighty five percent of the mutations with large effects on disease-related traits habor at exome. TES includes a certain set of target genes. Therefore it is much cheaper and faster than Sanger sequencing of each gene or whole exome sequencing and has deeper coverage. Therefore a numerous deafness samples could be screened through TES without substantial cost of time and money.4) TES has contributed to identification of novel genes causing hereditary hearing loss.9,10,11,12,13) The efficacy of TES has been reported from several studies.14,15,16)
Customized Hierarchical Molecular Genetic Test Protocol in Koreans
Based upon TES, we developed new diagnostic strategy combining phenotype-driven candidate gene approach and TES to find out the causative mutation of hearing loss. First, patients with certain phenotypic marker related to the hearing loss genes underwent Sanger sequencing to identify variants in corresponding candidate genes. Then GJB2 sequencing was performed for the remaining patients because the mutation in GJB2 was one of the most frequent mutation in NSHL cases.
Next, we performed TES. The number of candidate genes of TES and filtering steps were different according to the cases. In multiplex families, which has two or more NSHL members, 80 reported NSHL-related genes were screened (TES-80). After aligned to the human genome reference sequence (hg19), selected single nucleotide variations or indels were filtered through five steps which used the property of multiplex family. As a basic step, the variants with a low quality score (<20) were excluded. Then the variants, which did not coincide with the inheritance pattern of mulplex family, were discarded. Remained variants were checked in 80 normal hearing population and detected variants were excluded. Finally, segregation study in other families was performed by Sanger sequencing. Phenotype matching was also considered.
In sporadic severe to profound hearing loss families, 204 reported hearing loss-related genes were screened (TES-204). The results were aligned to the human genome reference sequence (hg19). Variants, which were detected in our in-house database composed of exomes of 81 Korean individuals, were excluded. Then low quality of genotyping (<30) and reads (<20) were discarded. The inheritance pattern of hearing loss was also checked. Lastly, the variants identified in our 276 Korean normal hearing control chromosomes were excluded.
Genetic Etiology in Korean Multiplex Families
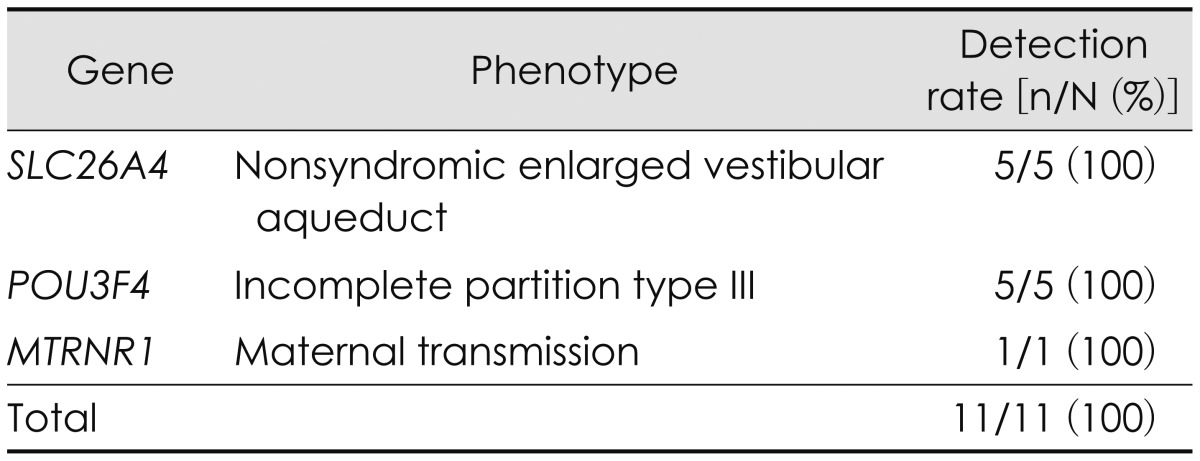
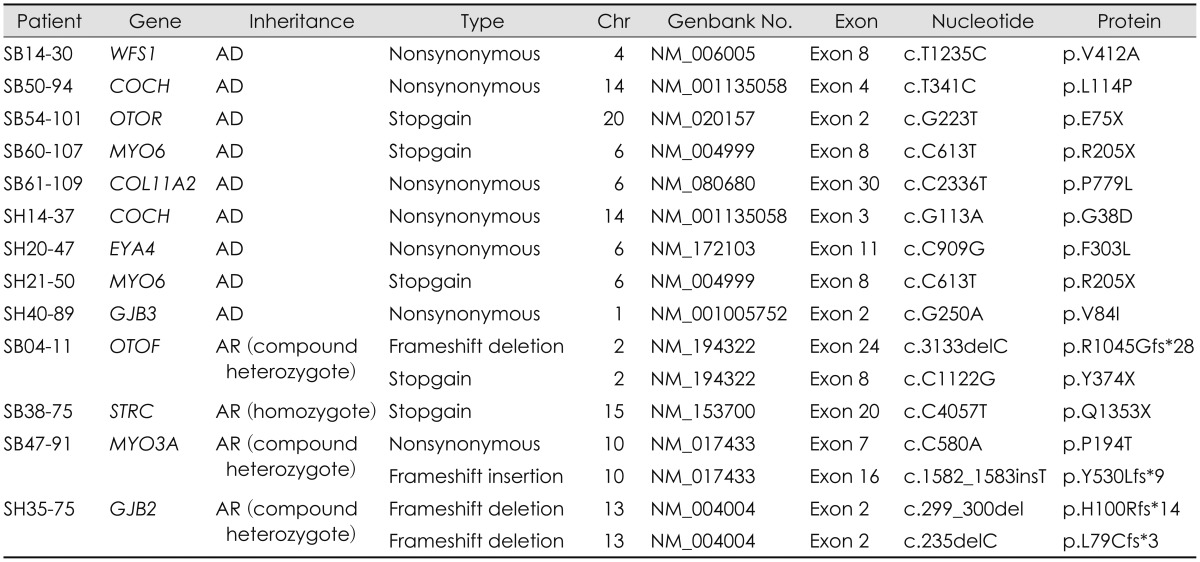
Our new diagnostic strategy was performed in 32 multiplex families to find out the causative mutations.16) Among 32 probands, 11 probands showed specific phenotype. Five probands showed bilateral enlarged vestibular aqueduct and another 5 probands showed incomplete partition type III on temporal bone CT. Each proband underwent Sanger sequencing of the corresponding candidate genes, SLC26A4 and POU3F4, respectively. In addition to 10 probands, one proband showed specific maternal inheritance pattern of hearing loss and mitochondrial DNA genes were sequenced. As a result, the causative mutations were found in these eleven families (Table 1). The remained 21 NSHL probands underwent GJB2 sequencing and 2 probands were identified to have known pathogenic mutations in GJB2 gene. Nest, TES of 80 reported NSHL-related genes (TES-80) was performed in 20 probands including one proband with mutations in GJB2 gene. Then five filtering steps, as mentioned above, were performed. Finally, among 20 probands, 13 probands found out their causative mutations (Table 2).
Consequently, among 32 multiplex families, the causative mutations were identified in 25 probands (78.1%) by our new diagnostic strategy combining phenotype-driven candidate gene approach and TES. Analyzing the results of all cases, molecular genetic diagnosis was completed in 9 (69.2%) form 13 autosomal dominant families and 9 (75.0%) from 12 recessive families. The causative gene of autosomal dominant families included WFS1, COCH, TECTA, MYO6, COL11A2, EYA4, and GJB3. The causative gene of autosomal recessive families included OTOF, STRC, MYO3A, GJB2, and SLC26A4. The causative mutations of remained families were one MRNR1 mutation with maternal inheritance and 5 POU3F4 mutation with X-linked inheritance.
Genetic Etiology in Korean Sporadic Severe to Profound Hearing Loss Families
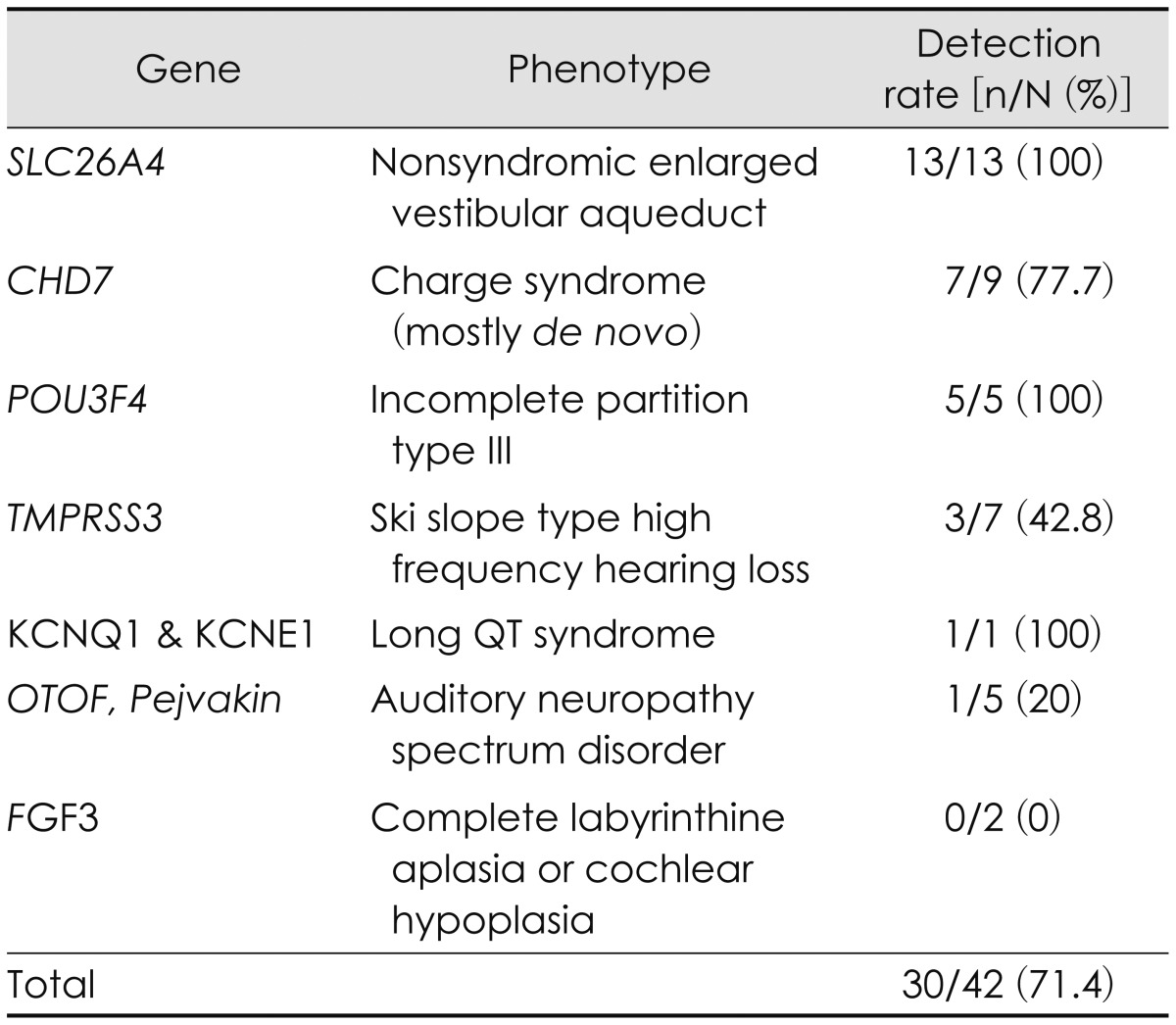
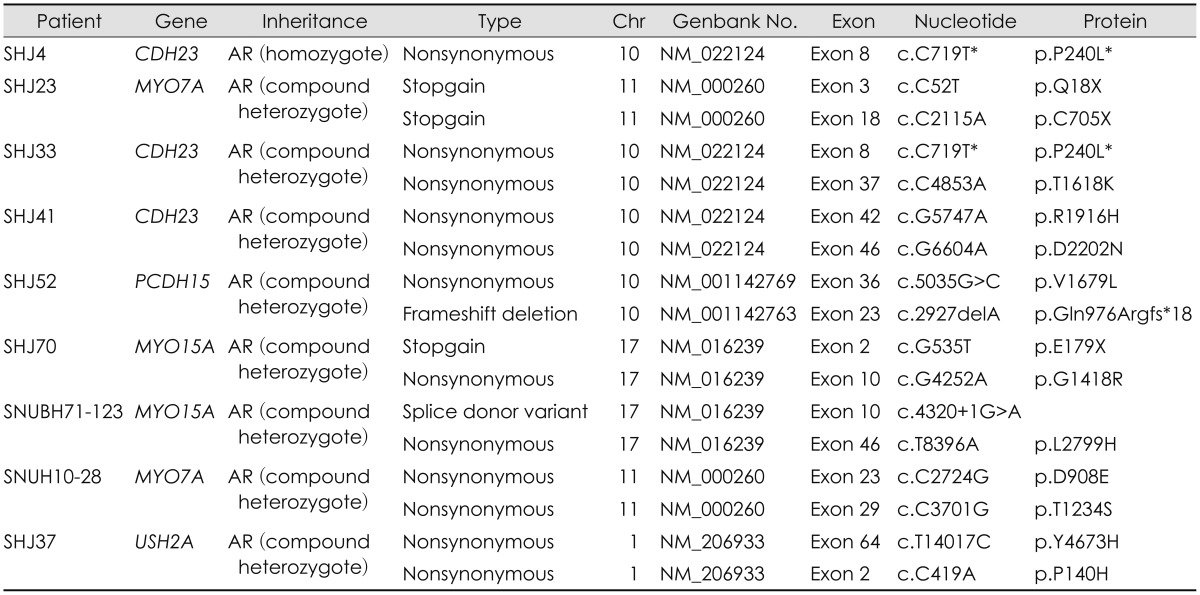
Our new diagnostic strategy combining phenotype-driven candidate gene approach and TES was performed in 93 patients with severe to profound SNHL to find out the causative mutation of hearing loss. Among 93 probands, 42 probands showed certain phenotypic marker related to the hearing loss genes and were subject to Sanger sequencing of related candidate genes. Thirty (71.4%) of 42 probands completed molecular genetic diagnosis successfully. Probands who showed enlarged vestibular aqueduct (n=13), incomplete partition type III (n=5) and long Q-T syndrome (n=1) were subject to direct sequencing of SLC26A4, POU3F4, and KCNQ1, respectively. The pathogenic variants were detected in all these probands. Other probands, who underwent direct sequencing of CHD7, TMPRSS3, and OTOF, showed the detection rate of 77.7%, 42.3%, and 20%, respectively. Therefore, enlarged vestibular aqueduct, incomplete partition type III, and long Q-T syndrome were the phenotype markers which predicted pathogenic variants most accurately (Table 3). Fifty one probands without phenotypic marker underwent GJB2 sequencing and the pathogenic variants were detected in 10 probands. As a result, the pathogenic variants were identified in 40 (43.0%) out of 93 probands through phenotype-driven candidate gene approach. Fifty three remained probands were subjected to TES-204. However, 8 probands rejected TES-204 and 45 probands underwent TES-204. The causative mutations were detected in 11 (24.4%) out of 45 probands. Nine out of 11 causative mutations were newly detected through TES-204 (Table 4). These causative mutations resided at CDH23 (n=3), MYO15A (n=2), MYO7A (n=2), ACTG1 (n=1), USH2A (n=1), PCDH15 (n=1), and MYO3A (n=1).
Consequently, the causative mutations were identified in 51 (54.8%) out of 93 sporadic severe to profound SNHL patients. At least 54.8% of sporadic severe to profound SNHL patients seemed to be monogenic Mendelian disorders in Koreans. The mutations in SLC26A4 and GJB2 were the most common causative mutations and responsible for 24.7% (23/93) of total patients.
Conclusions
In this paper, we demonstrated the value of new diagnostic strategy combining phenotype-driven candidate gene approach and TES for detecting causative mutations. The causative mutations in Korean familial NSHL and sporadic severe to profound SNHL patients were presented. Based upon the results, the value of this strategy as a diagnostic tool seems to be promising. Although whole genome and exome sequencing have advanced as the development of NGS, this new strategy could be a good screening and diagnostic tool to find the causative mutations.