Introduction
A congenital cholesteatoma (CC) is a pearl-shaped epidermoid cyst derived from the keratinizing squamous epithelium within the middle ear cleft. To distinguish a CC from an acquired one, several authors have revised the diagnostic criteria, and the revision of Levenson, et al.1) is generally accepted. According to this revision, a CC is a pearly white mass medial to an intact tympanic membrane, a normal pars tensa and pars flaccida, and no history of otorrhea, perforation, or previous otologic procedure, while a history of otitis media does not influence the diagnosis.
A CC may develop at five temporal bone sites: the petrous apex, cerebellopontine angle, middle-ear cavity, external auditory canal (EAC), and mastoid process. Of these, the mastoid process has the lowest incidence.2)
We reported a case of CC of mastoid origin in 2007;3) very few such cases have been reported worldwide. When we experienced a second case, we began to doubt the rarity of this entity. With a brief literature review, we consider how the cholesteatoma localizes to the tip of the mastoid bone.
Case Report
A 59-year-old man presented with a 5-month history of right-sided ear discharge. There was no history of ear discharge, perforation, previous otologic procedures, or other otological complaints involving the right ear.
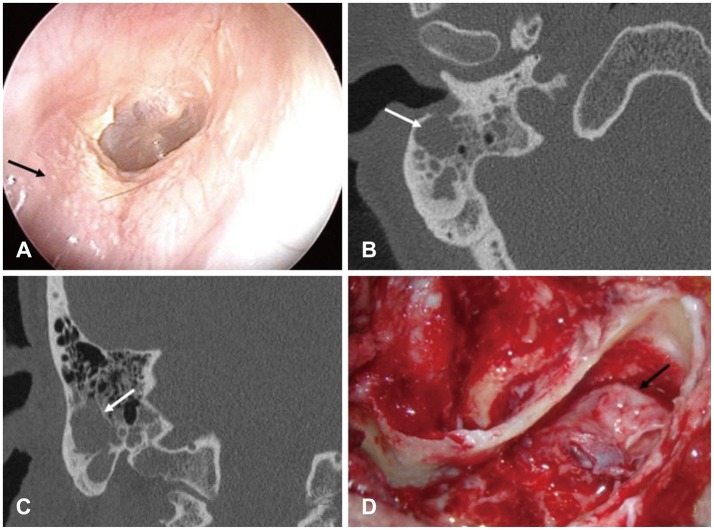
Physical examination revealed an intact tympanic membrane (Fig. 1A) and audiometric evaluation showed normal hearing. Computed tomography revealed a mass occupying the mastoid tip extending to the posteroinferior wall of the EAC (Fig. 1B, C). A simple mastoidectomy was performed. At surgery, the air cell in the mastoid tip area was filled with cholesteatoma and the posteroinferior wall of the EAC was partially destroyed (Fig. 1D). The cholesteatoma sac extended to the posterior wall of the EAC, but did not reach the mastoid antrum, middle ear cavity, or bony plate covering the sigmoid sinus. In this patient, Koerner's septum was well developed. The mucosa of the antrum was normal and the other air cells at the mastoid tip were filled with cholesterol granuloma. The cholesteatoma sac was completely isolated from the mastoid antrum and lateral air cell in the mastoid tip area. The sac was removed completely and the EAC was reconstructed using conchal cartilage.
On follow-up after 3 months, pure-tone audiometry indicated that the patient's hearing level was as same as before and there was no problem with the reconstructed EAC.
Discussion
Various hypotheses have been proposed to explain the pathogenesis of CC, which has been the subject of intense discussion for over a century. The main theories in this respect are: metaplasia, invagination, embryonal epithelial rest, and implantation.
The metaplasia theory is that non-squamous epithelium of the middle ear transforms into squamous epithelium in response to inflammation.4)
The invagination theory, also called the "stop signal theory", can be understood as the aberrant migration of external canal epithelium into the middle ear as a result of a developmental error. It is due to the failure to receive a stop signal from the tympanic ring, which acts as a 'barrier' in the migration of ectodermal tissue from the auditory canal to the middle ear.5) Tos6) reported a similar mechanism and proposed that after subsequent loosening of the retracted eardrum, some cells of the tympanic membrane become trapped in the middle ear cavity and eventually form an inclusion cholesteatoma.
The epithelial rest theory involves the abnormal persistence of epithelial rests. The theory was proposed by Teed7) in 1936 and is based on the findings in a 22-week-old human fetus in which an epidermoid (i.e., a distinct squamous cell nest) was found in the anterosuperior mesotympanum, the most common site for CC. In 1986, Michaels8) studied fetal temporal bones between the 10th and 33rd gestational week and hypothesized that the persistent growth of the epidermoid formation, rather than regression postpartum, could lead to CC in a few cases. Liang, et al.9) observed epidermoid formation in all four quadrants of the middle ear, the majority of which were in the anterosuperior quadrant. Also important was the observation of stratified squamous epithelium in the fetal middle ear in the early weeks after epidermoid formation.
Friedberg10) and House and Sheehy11) first suggested the implantation theory: the cause of CC is the implantation of squamous epithelium in the middle ear from either trauma or an unrecognized and subsequently healed tympanic membrane perforation. Northrop, et al.12) hypothesized that squamous epithelial cells in the amniotic fluid enter the anterosuperior mesotympanum via the eustachian tube, spread to the middle ear cavity, and the debris converts into a cholesteatoma.
A number of different studies have examined the etiopathogenesis of CC, and the four main theories all seem reasonable at times, but not at others. Is it possible that one of these four theories explains the etiology of our case, i.e., a CC localized to the tip area of mastoid bone?
First, the metaplasia theory is not likely to be applicable to CC of the mastoid tip. Friedberg10) noted that metaplasia would occur randomly in the middle ear cavity. However, it is difficult to imagine that a patient with a well-demarcated cholesteatoma at the mastoid tip has a normal middle ear cavity.
Second, the invagination theory is good for explaining lesions in the posteroinferior region of the middle ear, in proximity to the site of the embryonic tympanic ring. When contemplating CC of the mastoid tip, this is plausible. Aimi5) postulated that while the tympanic ring, which is found at the junction of the first and second brachial arches in a 10-week fetus, was absent, a 16.5-week fetus showed significant ectodermal tissue projection into the mesenchyme. The mastoid process does not start to develop until birth and is located in the posterior part of the tympanic ring at birth. It is possible that ectodermal tissue could migrate to the mastoid process during fetal life and grow there after birth. Although possible, it is extremely improbable.
Third, the epithelial rest theory is good for explaining lesions of the anterosuperior region of the lateral wall of the tympanic cavity where an epidermal structure is traditionally found. Epidermoid formation (i.e., a potential precursor of CC) originates from the ectodermal tissue of the first brachial cleft during development of the EAC. If an epidermoid formation occurred in the undeveloped mastoid during fetal life and grew there after birth as a precursor of CC, then this theory could explain a CC of the mastoid tip. However, epidermoid formation has never been reported in the mastoid and, even if possible, would be extremely rare.
Fourth, the implantation theory is a possible, rare cause of CC. It can account for lesions isolated to many different sites, such as the mastoid and cranial areas. Madeline and Elster13) suggested that cranial suture development is a series of complex steps from early embryogenesis to the third decade of life. Canalis, et al.14) suggested that a cranial cholesteatoma could arise from squamous epithelium trapped within the suture at some time during closure of the mastoid fontanel; during suture closure, the persistent patency of the fontanel may allow the possibility of trapping of ectodermally derived squamous cells, as in the process of simple sutures. In this regard, such entrapment of squamous cells, and the formation of a cholesteatoma as a result, would also be plausible for any cranial suture.
An intradiploic epidermoid cyst, more commonly described in the neurosurgical literature, is regarded as a congenital implantation cholesteatoma. It is indistinguishable from a cranial cholesteatoma histologically and radiologically. Clark, et al.15) proposed that a CC and intradiploic epidermoid cyst are one and the same condition. All three conditions (i.e., congenital cholesteatoma localized only to mastoid bone, cranial cholesteatoma, and intradiploic epidermoid cyst) are extremely rare and we believe that there is no better way to explain their origins than the implantation theory. The epicenter in our case was thought to be the tympanomastoid suture, which was localized to the tip of the mastoid bone. It did not extend to the mastoid antrum and destroy Koerner's septum. The implantation theory is the best explanation for our case.
The origin of CC continues to be controversial. There are four competing hypotheses, which all seem plausible. It is likely that no single mechanism explains the origin of all CCs, and the implantation theory seems best for explaining our case.
Since first report of a CC of mastoid origin in 1965, eight more papers have reported a total of 16 cases.3) This entity was once regarded as extremely rare; however, in the last 10 years, five more publications have reported 13 more cases, so this entity may not be that rare.
We reported a CC of mastoid origin, focusing on its rarity, in 2007.3) In that case, the cholesteatoma destroyed the tegmen mastoideum, the bony plate of the posterior fossa, and the bony plate covering the sigmoid sinus, but the middle ear and aditus ad antrum were clear. By contrast, in the current case, the cholesteatoma was solely located at the tip of the mastoid bone and even Koerner's septum was well developed. To broaden our knowledge of CC, this case is good for explaining the etiology of CC because it is so far from the predictable trajectory. Among the four competing theories on the pathogenesis of CC, the implantation theory best explains our case and other similar entities (i.e., cranial cholesteatoma and intra-epidermoid cyst).